为什么说 AlphaFold 2 足以改变全人类?
以下文章来源于夕小瑶的卖萌屋 ,作者炼丹学徒
夕小瑶的卖萌屋
有妹子、有猛料的人工智能前沿算法故事会。关键词:自然语言处理、计算机视觉、深度学习、推荐系统、搜索、计算广告、机器学习
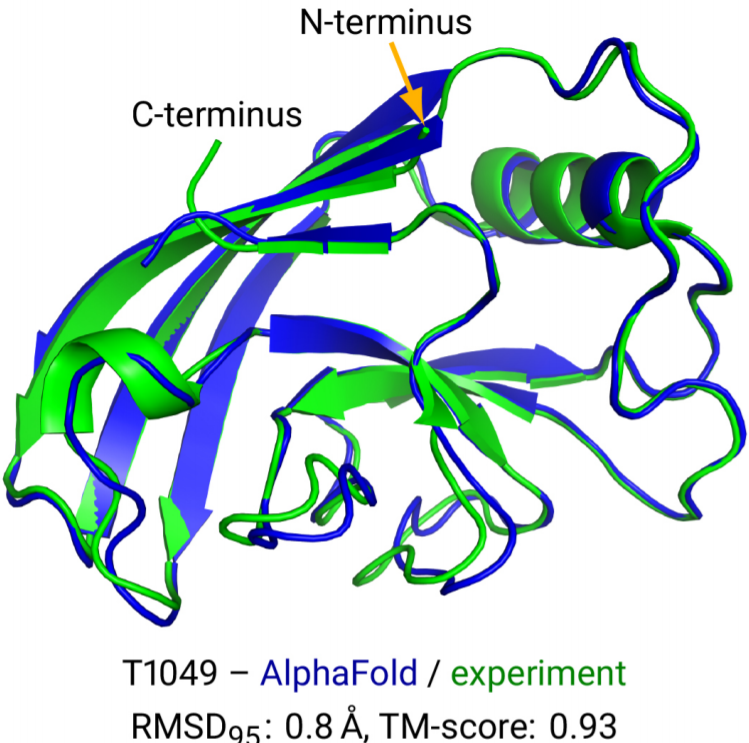
Highly accurate protein structure prediction with AlphaFold
https://github.com/deepmind/alphafold
https://www.nature.com/articles/s41586-021-03819-2_reference.pdf
1
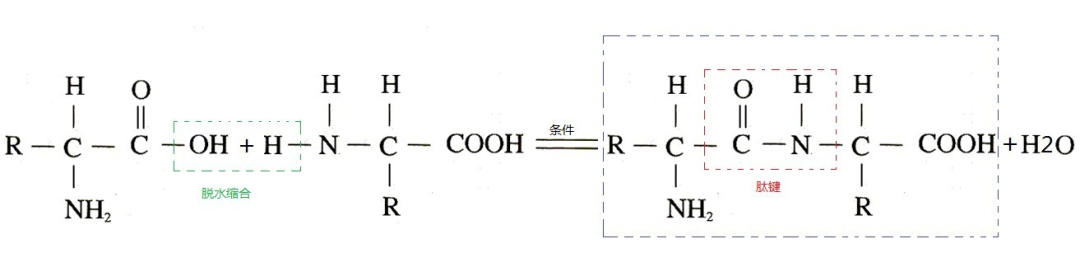
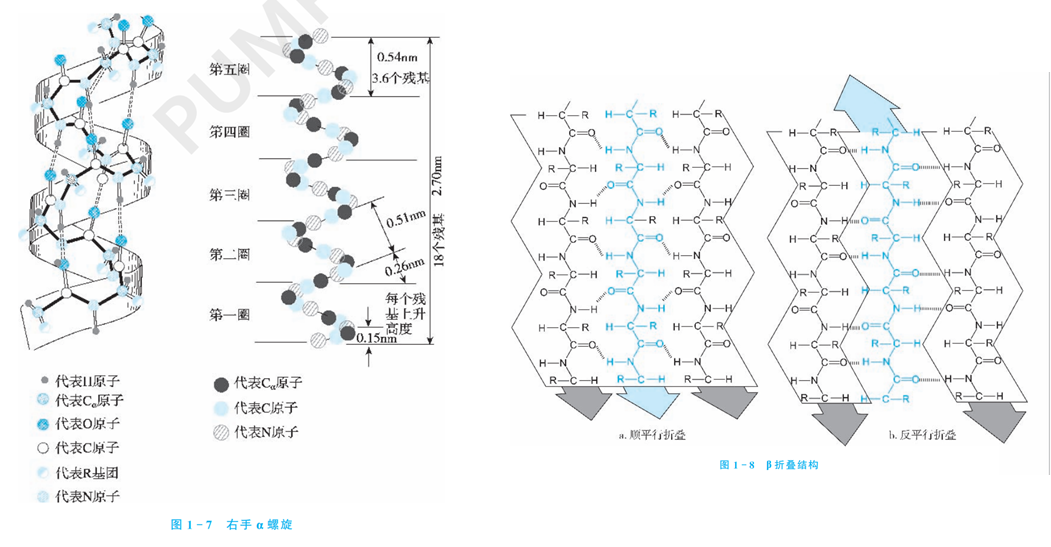
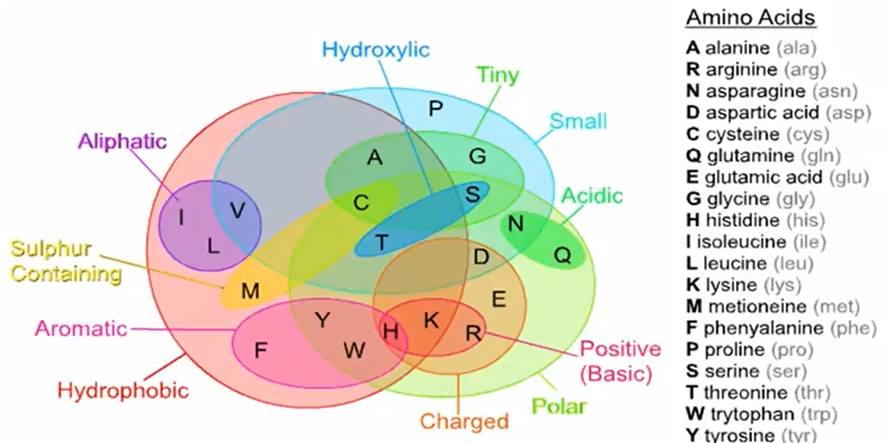
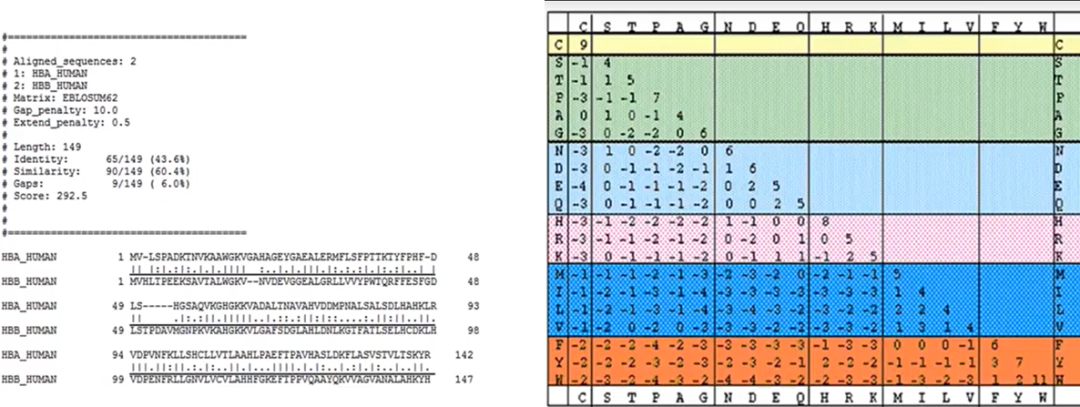
基础知识

2
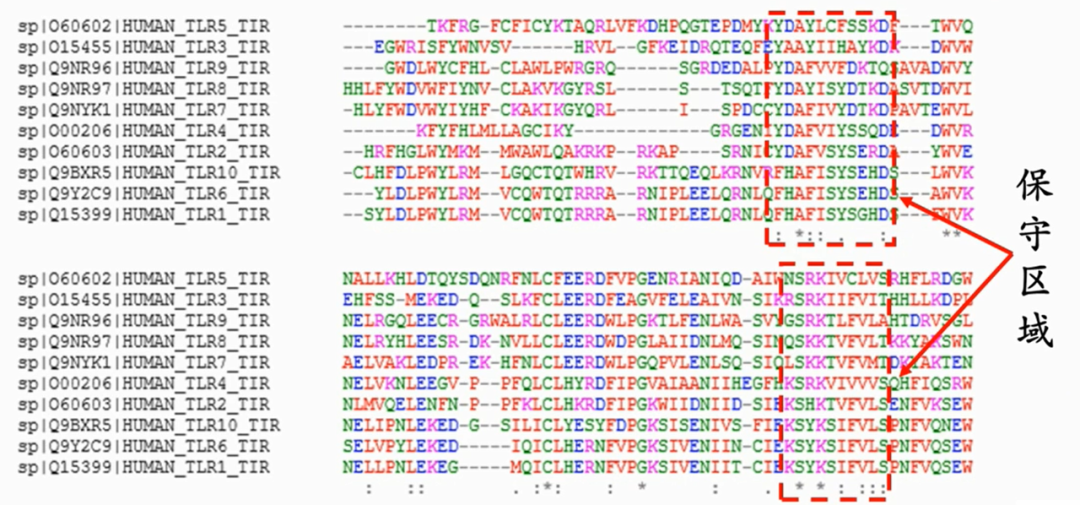
为啥深度学习研究蛋白质结构很重要
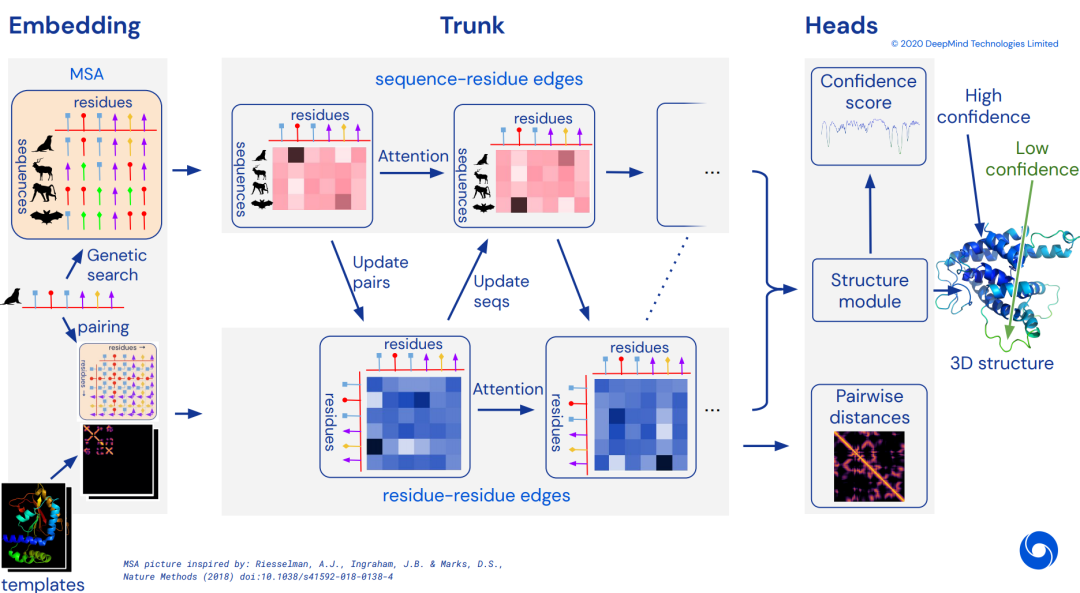
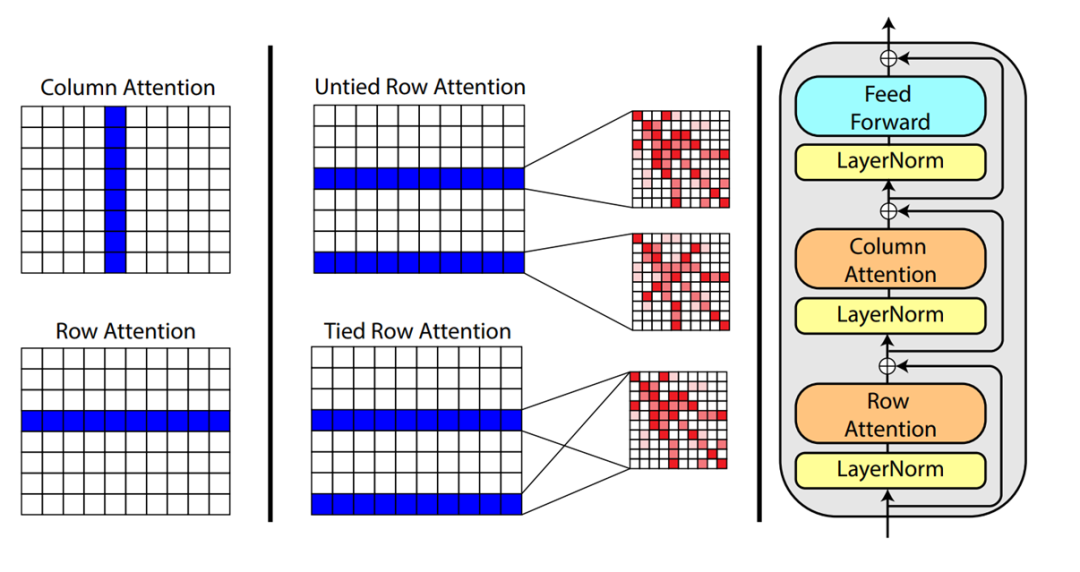
3
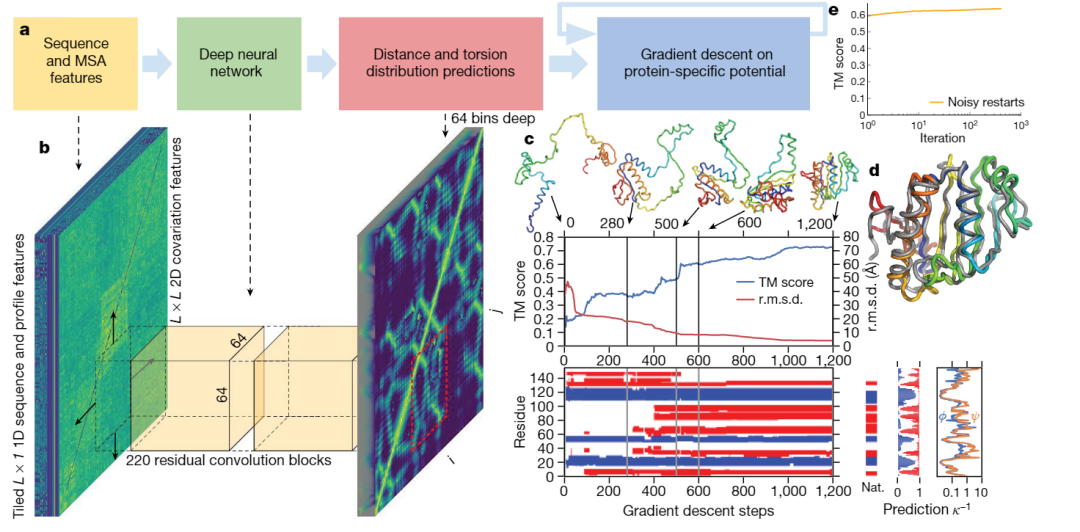
AlphaFold2
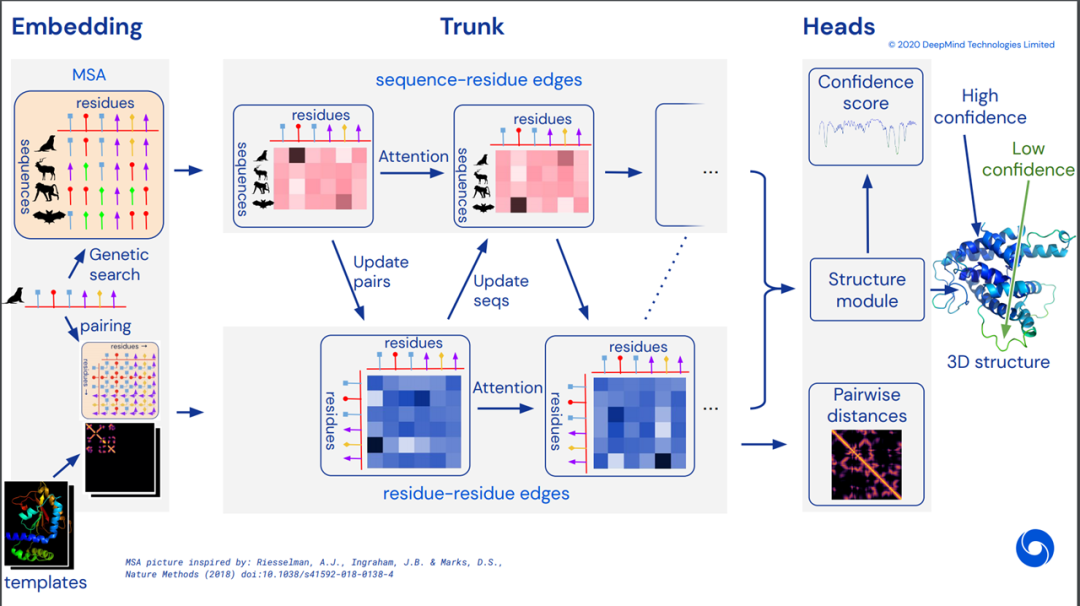
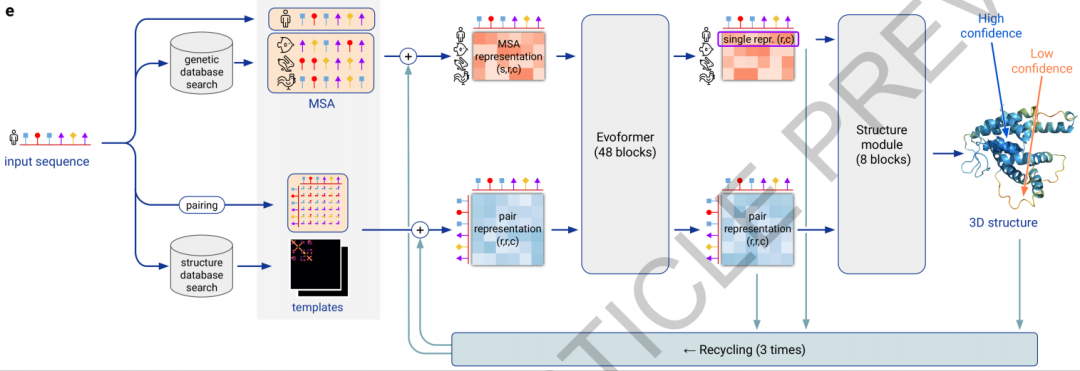
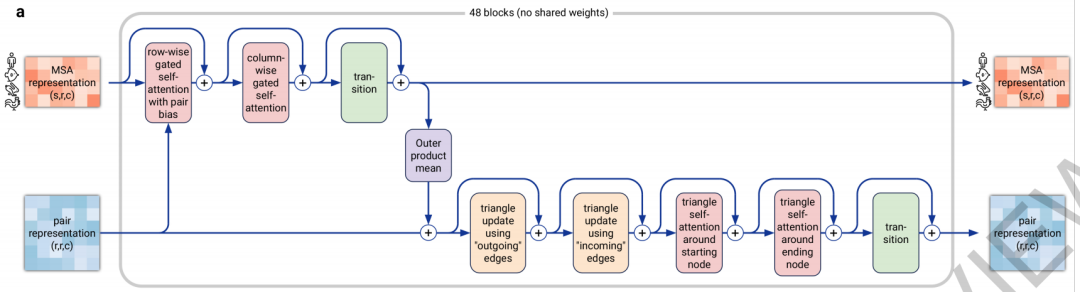
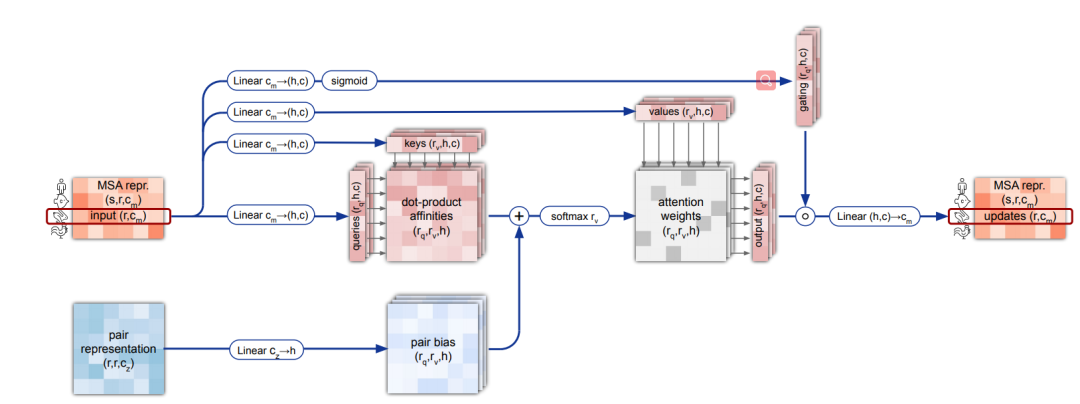
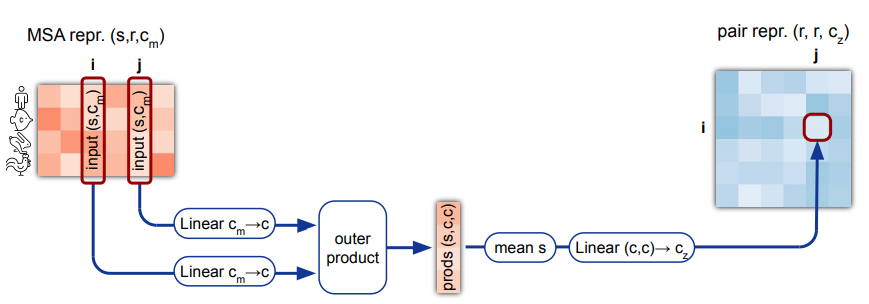
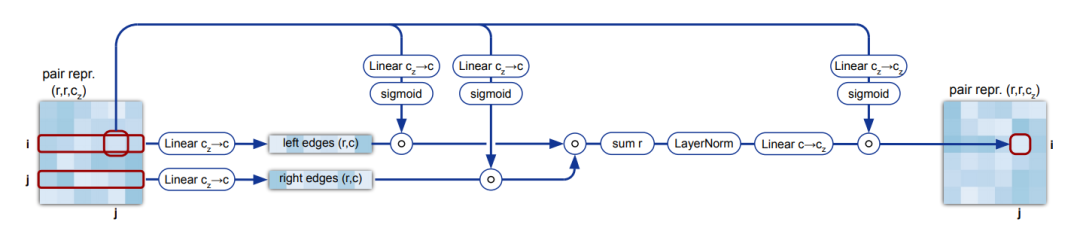
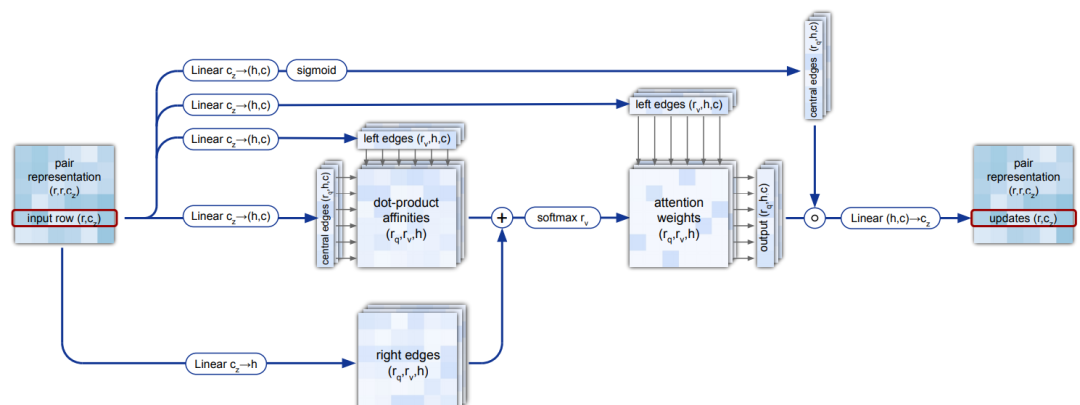

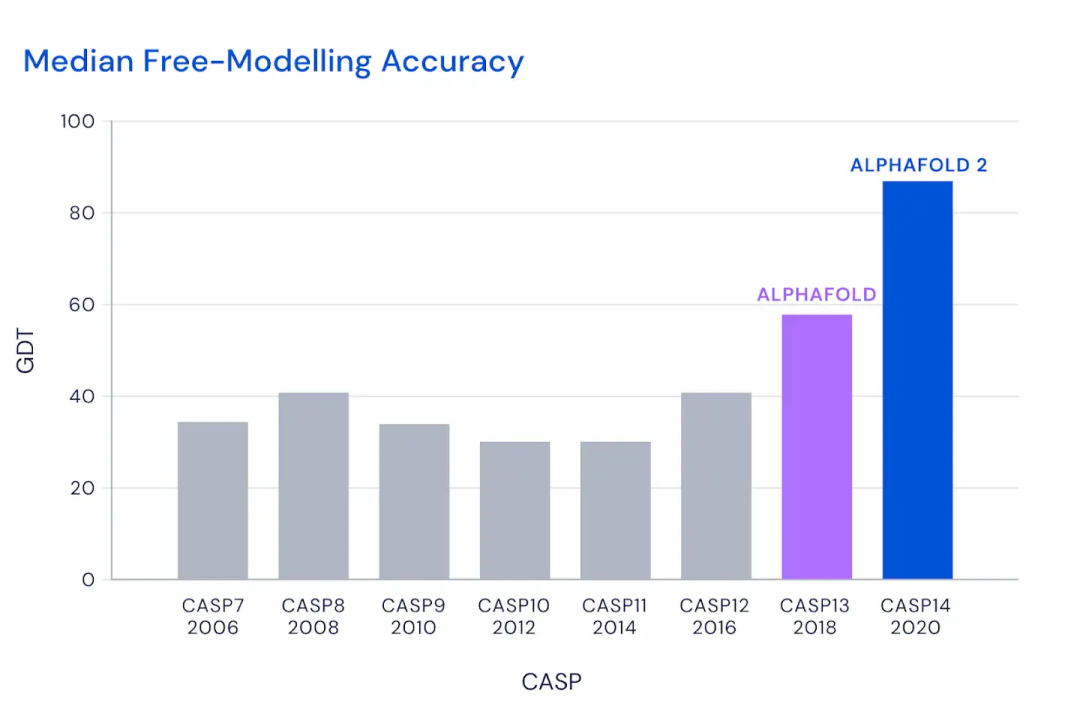
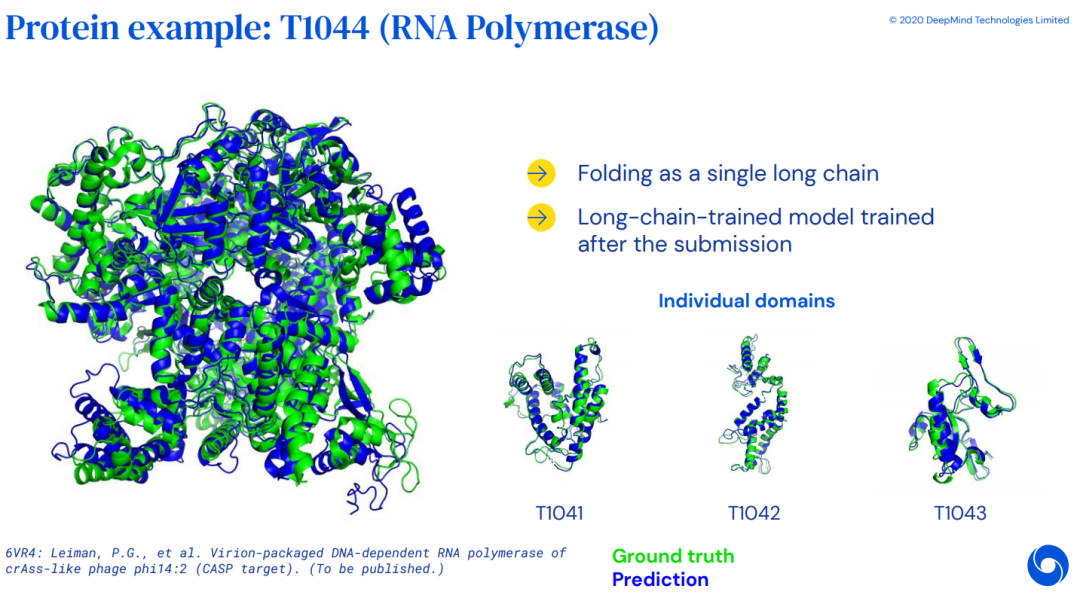
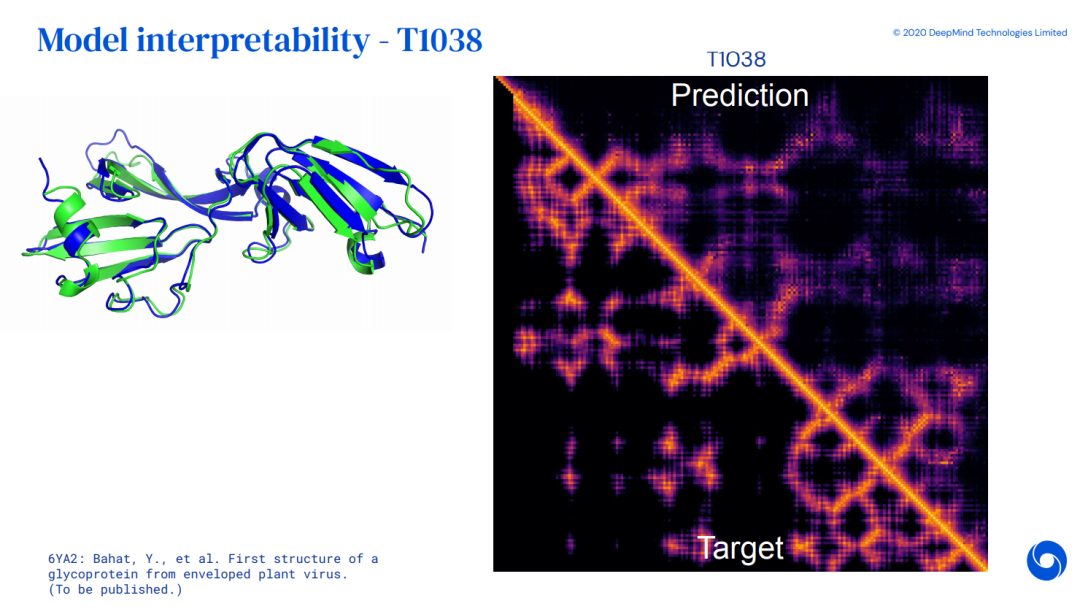
4
总结和展望
AI科技评论
聚焦AI前沿研究,关注AI青年成长
公众号